По материалам статьи
С.О. Тебердиева, Л.В. Ушакова, Е.А. Филиппова, В.Г. Быченко, Е.И. Дорофеева, А.А. Буров, Д.Н. Дегтярев
ФГБУ «Научный центр акушерства, гинекологии и перинатологии им. академика В.И. Кулакова» Минздрава России, Москва, Россия Diagnostic value of neuroimaging techniques in newb
В структуре младенческой смертности аномалии развития занимают третье место. Современные успехи реаниматологии, неонатологии и детской хирургии в последние десятилетия определили возможность оказания неотложной помощи новорожденным детям с тяжелыми пороками развития. Тяжесть состояния таких детей при рождении зачастую обусловлена легочно-сердечной недостаточностью, нестабильной гемодинамикой, что повышает риск перинатального поражения ЦНС. Традиционно в качестве основного метода ранней диагностики структурных поражений головного мозга у новорожденных детей используется нейросонография. Широкое применение этого метода связано с общедоступностью, безопасностью и малой инвазивностью. Метод бесценен в диагностике поражений головного мозга у детей, находящихся в отделении реанимации, транспортировка которых для проведения магнитно-резонансной томографии (МРТ) невозможна в связи с тяжестью состояния.
Более точная информация о наличии и характере структурных изменений ЦНС может быть получена на основании МРТ. Помимо более высокой степени детализации анатомических структур головного мозга, этот метод характеризуется отсутствием лучевой нагрузки на организм, возможностью исследования в различных плоскостях, не перемещая пациента, и высоким тканевым контрастом. Кроме того, МРТ является стандартизованным воспроизводимым методом, что делает его более объективным по сравнению с другими видами нейровизуализации, включая нейросонографию. Недостатком метода является необходимость специальной подготовки пациента (транспортировка, дополнительная медикаментозная седация, необходимость магнит-совместимого кувеза для критически тяжелых новорожденных и др.) и существенно более высокая стоимость. Как правило, показанием к данному исследованию является неврологический статус ребенка в совокупности с данными лабораторно-инструментальных методов обследования. Накопленный в Научном центре акушерства, гинекологии и перинатологии имени В.И. Кулакова опыт дополнительного обследования новорожденных в пред- и послеоперационном периодах позволяет выяснить частоту структурных поражений ЦНС у детей с врожденными пороками внутренних органов и уточнить клиническую информативность каждого из используемых методов.
Цель исследования: установить частоту и характер поражений головного мозга методами нейровизуализации, определяющими транзиторные и необратимые поражения функции ЦНС у новорожденных детей с пороками развития внутренних органов в периоперационном периоде.
Характеристика детей и методы исследования
Для достижения поставленной цели проанализированы 243 истории болезни детей с врожденными пороками внутренних органов. Все дети были рождены в Научном центре акушерства, гинекологии и перинатологии имени академика В.И. Кулакова в период с 14 января 2014 г. по 23 февраля 2015 г. Из 243 новорожденных 211 (87%) были доношенными (37– 41 (38,4+0,9) нед), 32 (13%) – недоношенными (32– 36 (35,2+1,1) нед). Распределение по полу было следующим: 117 (48%) девочек и 126 (52%) мальчиков. Дети родились с оценкой по шкале Апгар от 4 до 9 (7,2+1,8) баллов на 1-й минуте жизни и от 5 до 9 (8,3+2,0) баллов на 5-й минуте жизни, 16% (n=37) детей испытывали асфиксию средней степени тяжести при рождении, тяжелую асфиксию перенесли 0,8% (n=2) новорожденных.
На основании данных пренатальной диагностики сразу после рождения детей переводили в отделение хирургии, реанимации и интенсивной терапии новорожденных для углубленного обследования, и хирургического лечения.
Наиболее часто поступали в отделение дети с пороками развития почек (24%), на втором месте по частоте – порок развития передней брюшной стенки – гастрошизис (15%), на третьем месте – пороки развития легких и киста яичника (по 11%). Остальные врожденные пороки встречались реже. Более 80% (n=197) детей требовалась хирургическая коррекция врожденных пороков развития, из них 36% (n=71) были оперированы в первые 4–8 ч от момента рождения, 22% (n=43) – к концу 1-х суток жизни. Остальные дети были оперированы в период с 2-х до 18-х суток жизни.
В связи с поставленной целью выявления факторов риска перинатального поражения ЦНС в дооперационном, интраоперационном и послеоперационном периодах всем детям оценивался неврологический статус, проводилось скрининговое нейросонографическое исследование (трехкратно) и нейрофизиологические методы исследования (ЭЭГ, амплитуд-интегрированная ЭЭГ – аЭЭГ). В соответствии с внутренним протоколом отделения нейросонографию осуществляли в первые часы после рождения (4,9±1,7 ч), перед оперативным вмешательством, через 24–48 ч после оперативного вмешательства и далее не реже 1 раза в неделю. Исследование проводили врачи отделения ультразвуковой (УЗ) диагностики в неонатологии и педиатрии на УЗ-аппарате экспертного класса SIEMENS Acuson S2000.
Неврологический осмотр обследуемых больных проводился согласно стандартизованным методикам с учетом гестационного возраста ребенка (Бондаренко Е.С., Журба Л.Т., Мастюкова Е.А., Dubwits L.M.S., Prechtl H.F.R., Шабалов Н.П., Пальчик А.Б.) до операции и в послеоперационном периоде после отмены обезболивающих средств. Неврологический статус оценен у 16% больных в дооперационном периоде и у 54% – после хирургического лечения. Мониторинг церебральной функции (аЭЭГ) выполнен 15 (6,2%) детям в непрерывном режиме в течение 24 ч аппаратом Natus Olimpic Brainz Monitor. У 8 (3,3%) детей были обнаружены изменения паттерна аЭЭГ – проведена стандартная ЭЭГ аппаратом Энцефалан-ЭЭГ 19/26 Медиком МТД.
МРТ-исследование было проведено у 15 (6,2%) из 243 новорожденных c врожденными пороками внутренних органов. В том числе 4 ребенка страдали врожденной диафрагмальной грыжей, по 2 ребенка – гастрошизисом, гидронефрозом, стенозом двенадцатиперстной кишки, множественными врожденными пороками развития, по 1 ребенку – аденоматозом легкого, бронхогенной кистой, омфалоцеле. Исследование осуществляли на аппарате Siemens Magnetom Verio с индукцией магнитного поля 3 Tл, оснащенном специализированной педиатрической катушкой для обследования головного мозга, системой контроля жизненно важных функций (SpO2, ЧCC) и МР-совместимым аппаратом искусственной вентиляции легких. МРТ головного мозга проводилась в режимах: T1, T2, DWI, FLAIR, SWI, времяпролетной магнитно-резонансной ангиографии (МР-АГ), в том числе у 2 детей в режиме контрастной МР-АГ.
Результаты
По результатам комплексного неврологического и клинико-инструментального обследования детей с врожденными пороками внутренних органов более 42% (n=102) имели признаки поражения ЦНС. Основными проявлениями перинатального поражения ЦНС были: синдром угнетения – у 34 (33%), изменение мышечного тонуса и двигательные нарушения – у 59 (58%), судорожный синдром – у 6 (6%), что и соответствовало результатам, полученным при использовании методов нейровизуализации, аЭЭГ и ЭЭГ. Получилось, что в раннем и позднем постоперационном периодах, а также перед выпиской синдром двигательных нарушений регистрировался у 8–10 (8–9%) детей. Синдром мышечной дистонии устанавливался неврологом чаще всего перед выпиской и наблюдался у 38 детей. Гипертензионно-гидроцефальный синдром был выявлен в постоперационном периоде у 3 детей с врожденной диафрагмальной грыжей и потребовал вентрикулоперитонеального шунтирования. Судорожный синдром до операции на блюдался у 1 (1%) ребенка, после операции – у 6 (6%) новорожденных с трансформацией в диагноз эпилепсии у 2 (2%) детей.
Наиболее тяжелые неврологические проявления гипоксически-ишемической энцефалопатии имели дети с врожденной диафрагмальной грыжей. Энцефалопатия у этих детей сочеталась с высокой легочной гипертензией, повышенным давлением в системе верхней полой вены, что влияло на церебральную гемодинамику и ее ауторегуляцию.
Хирургическая коррекция таких врожденных пороков, как гастрошизис и омфалоцеле, приводит к повышению внутрибрюшного давления и сказывается на гемодинамике в системе нижней полой вены и аорты, а также влияет на почечный кровоток.
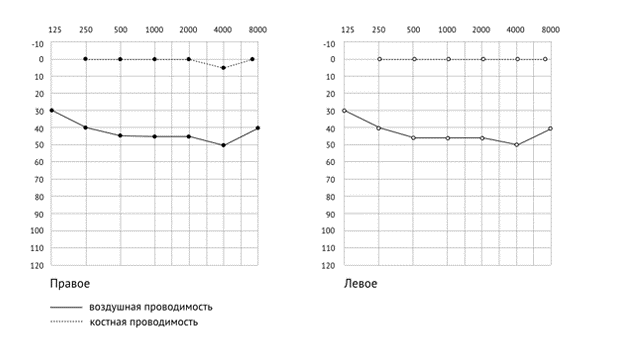
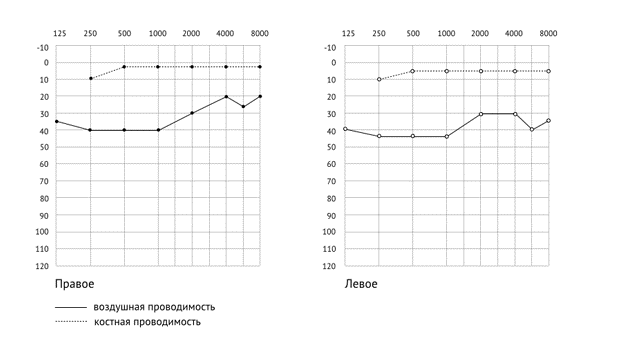
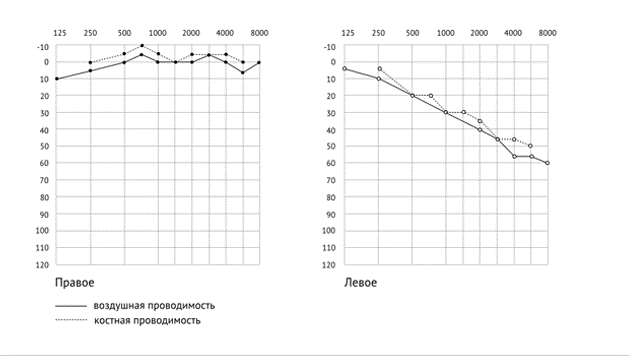
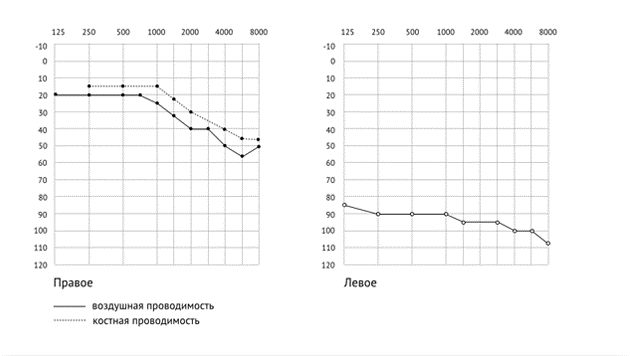
У 50% детей с признаками поражения ЦНС, по данным нейросонографии, были выявлены различные структурные изменения головного мозга. Из них наиболее часто встречалось расширение наружных и внутренних ликворных пространств, внутрижелудочные кровоизлияния различной степени тяжести.
МРТ головного проводилась 15 из 53 детей с выявленными при нейросонографии структурными поражениями ЦНС. По данным МРТ, внутрижелудочковые кровоизлияния обнаружены у 8 (3,3%) из 243 пациентов с пороками развития внутренних органов, подоболочечные (субдуральные и субарахноидальные) кровоизлияния – у 3 (1,2%), церебральные кисты – у 1 (0,4%), паренхиматозные кровоизлияния – у 2 (0,8%). Тромбоз церебральных синусов методом МРТ был выявлен у 3 (1,2%) детей, у 1 (0,4%) ребенка имелись косвенные признаки этого нарушения по данным нейросонографии. Расширение наружных и внутренних ликворных пространств в сочетании с внутрижелудочковыми кровоизлияниями различной степени тяжести, расширением субдурального пространства наблюдалось чаще в группе детей с врожденной диафрагмальной грыжей – у 15 (20%). Замедленный кровоток и перегрузка системы верхней полой вены у 3 детей осложнились внутричерепной гипертензией, внутрижелудочковыми кровоизлияниями различной степени тяжести, церебральным синус-тромбозом, структурным повреждениям головного мозга с клиническими проявлениями гипоксически-ишемической энцефалопатии, определяя в дальнейшем неврологический исход. Острое нарушение мозгового кровообращения по ишемическому типу в бассейне правой задней мозговой артерии с поражением правой таламической области было выявлено у 1 ребенка с врожденной диафрагмальной грыжей в сочетании с коарктацией аорты (подтверждено по данным и нейросонографии, и МРТ).
Выводы В результате комплексного неврологического и клинико-инструментального обследования детей с врожденными пороками внутренних органов более чем у 40% установлены клинические признаки поражения ЦНС, которые в большинстве случаев – у 55 (54%) детей имели транзиторный характер, а у 22 (22%) – и органическую природу и сочетались со структурными изменениями головного мозга. Наиболее серьезные неврологические нарушения отмечались у детей с врожденной диафрагмальной грыжей, гастрошизисом, омфалоцеле, множественными врожденными пороками, тяжелыми пороками развития легких в связи с развивающимися гемодинамическими расстройствами, влияющими на мозговой кровоток.
Для полноценной оценки факторов риска дети с врожденными пороками нуждаются в амбулаторном катамнестическом наблюдении с оценкой психомоторного развития. Нейросонография и МРТ являются взаимодополняющими методами диагностики врожденных и перинатальных поражений головного мозга.
Материал подготовила врач ультразвуковой диагностики
Литвинова Г.В.